

This page provides a full description of the technology used for predicting breast cancer by applying deep learning to linked health records and mammography images, presented at https://pubs.rsna.org/doi/abs/10.1148/radiol.2019182622 . To cite the paper, please use: Akselrod-Ballin, A., Chorev, M., Shoshan, Y., Spiro, A., Hazan, A., Melamed, R., Barkan, E., Herzel, E., Naor, S., Karavani, E. and Koren, G., 2019. Predicting breast cancer by applying deep learning to linked health records and mammograms. Radiology, 292(2), pp.331-342.
The technology below is a result of a joint work between Maccabi Health Services, Assuta Medical Centers, and IBM Research.
Authors: Ayelet Akselrod-Ballin PhD, Michal Chorev PhD, Yoel Shoshan, Adam Spiro PhD, Alon Hazan Msc, Roie Melamed PhD, Ella Barkan Msc, Esma Herzel Msc, Shaked Naor Bsc, Ehud Karavani Bsc, Gideon Koren MD, Yaara Goldschmidt PhD, Varda Shalev MD MPH, Michal Rosen-Zvi PhD, Michal Guindy MD MPH
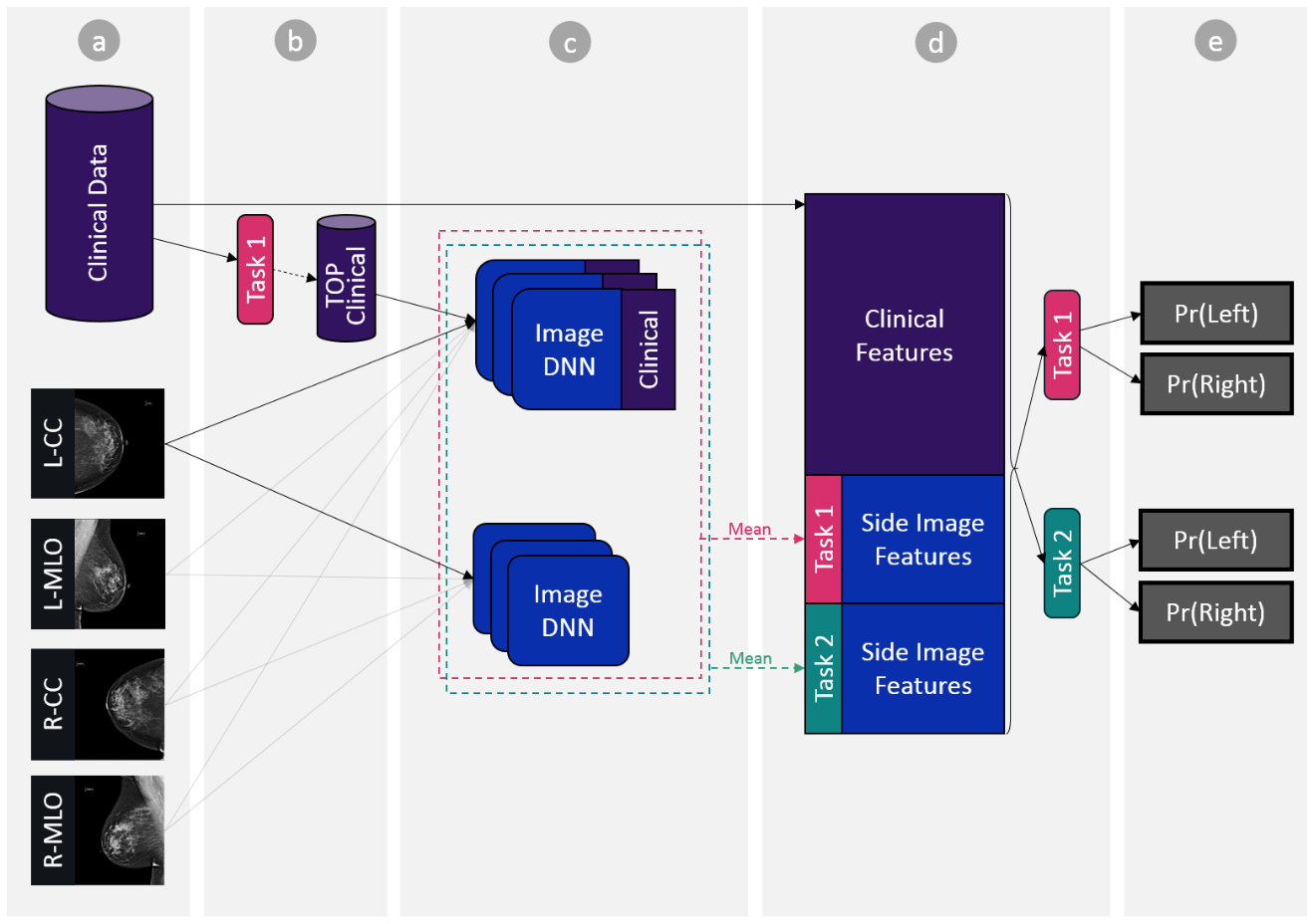
The above figure provides an illustration of the machine- and deep-learning algorithm that is trained on a linked dataset of mammography and detailed electronic health records. It achieves accuracy comparable to radiologists as defined by the American benchmark for screening digital mammography, performs in the acceptable range of radiologists, reveals additional clinical risk features, and reduces the likelihood of breast cancer misdiagnosis.
The Algorithm Pseudocode
Training
Step a: Data Preparation
Clinical data
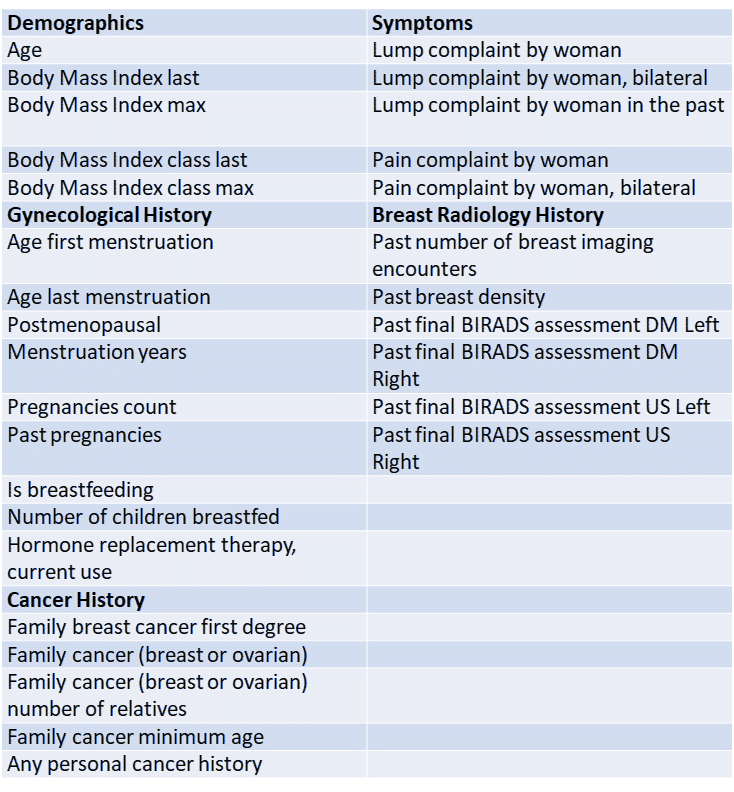
Prepare clinical data matrix “ClinicalFeaturesMat_FULL” of shape (M x N) – M denoting the number of patient study examples and N denoting the number of clinical features.
Important: Clinical features that include information regarding the mammogram read outcomes (such as BIRADS, biopsy result, etc.) are excluded from the list of clinical features.
Medical imaging data
For each patient study use standard four-view images (LCC, LMLO, RCC, RMLO) and discard others if exist. If multiple images exist for the same side-view, for example, more than one LCC images in a study, use the latest image.
Step b: Feature Selection
Build matrix “MalignancyLabels” of shape (M, 1) – M denoting the number of patient study examples. This matrix will map between patient study to the ground truth cancer-positive biopsy label.
# train a gradient boosting based model gb_model = xgboost.train(ClinicalFeaturesMat_FULL, MalignancyLabels) # select top features T <-number of features desired For each feature f in features Scores[f] <- xgboost weight-based feature importance score(f) features <- features.sort(scores,’descending’) top_features <- features[0:T-1]
Build a new data matrix named “ClinicalFeaturesMat_TOP” of shape (M, T). M denoting the number of patient study examples and T denoting the number of top selected features.
In our case we started with 1343 clinical features which was reduced to T=30, see top features description below.
Here, we have tested the use of Gail’s features. However, the AUC obtained for Gail’s was inferior to the one obtained for the 30 features we have selected (AUC 0.54 vs. 0.63, p<.001).
Step c: Training Deep Learning Models
Train multiple models to be later used as an ensemble, see Pseudo Code below:
func_train_split(train_data, classification_task, use_clinical_features): model <- random start weights For each epoch For each sample in train_data, For image in images: If use_clinical_features Input = [image, top_clinical] Else Input = [image] model->forward(input) model->backward() optimizer->step() func_train(train_data): # task-1 = malignant vs. (benign or normal) # task-2 = (malignant or benign) vs. normal splits <- random train-validation from train_data splits (each 80% train %20 validation) For each classification_task in [task-1, task-2] For each split in splits: For each use_clinical_features in [‘yes’,’no’] tr, vl <- train_data.split(80,20, 'random') model[classification_task, split, use_clinical_features] <- func_train_split(tr, classification_task, use_clinical_features)
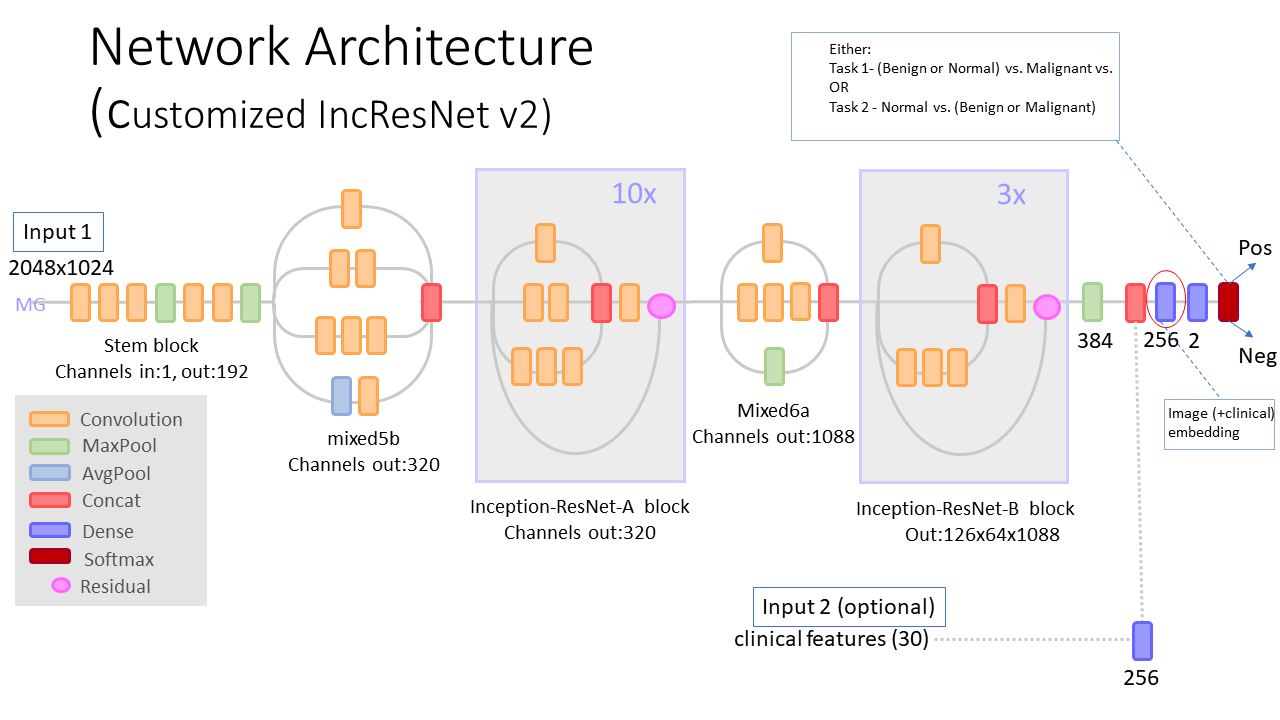
See model architecture diagram below.
Step d: Patient Vector Representation
Build encoding matrix “PatientBreastSideEncoding” of size [S x F]. S denoting number of breast sides and F the size of the total breast side encoding size.
Concatenate final breast side representation encoding vector. See Pseudo code below:
SET PatientBreastSideEncoding to empty matrix For each pat_study_side SET curr_encoding to empty vector curr_encoding.concat( clinical features from ClinicalFeaturesMat_FULL for relevant patient study side) For each task in [task-1, task-2] #per classification task SET avg_models_embedding <- 0 vector For each deep_learning_model trained on task avg_models_embedding += deep_learning_model.extract_features(pat_study_side) #features layer marked in red in diagram avg_models_embedding = avg_models_embedding / models_num curr_encoding.concat(avg_models_embedding) # Px,task-k denoting averaging deep learning models predictions over x, # w.r.t task-k curr_encoding.concat( Pcc,task ) curr_encoding.concat( Pmlo,task) curr_encoding.concat( Pcc,task) curr_encoding.concat( Pcc,task / (Pcc,task + Pmlo,task) ) curr_encoding.concat( Pmlo,task / (Pcc,task + Pmlo,task) ) curr_encoding.concat( | Pcc,task - Pmlo,task |) curr_encoding.concat( Max(Pcc,task, Pmlo,task) ) PatientBreastSideEncoding.concat(curr_encoding)
Step e: Final Model
- Depending on the final desired task, build either:
- Build matrix “Labels_Task_1_PerSide” of shape (S, 1) – S denoting the number of patient study breast sides examples. This matrix will map between patient study breast side to the ground truth of task 1
- Build matrix “Labels_Task_2_PerSide” of shape (S, 1) – S denoting the number of patient study breast sides examples. This matrix will map between patient study breast side to the ground truth of task 2
OR
# reminder: # task-1 = malignant vs. (benign or normal) # task-2 = (malignant or benign) vs. normal # Train a gradient boosting model using the vector representation # described in previous step. final_gb_model = xgboost.train(PatientBreastSideEncoding, Labels_Task_K_PerSide) #K being the selected task index
Testing
Step a: Data Preparation
Clinical data
Prepare clinical data test matrix “ClinicalFeaturesMat_FULL” of shape (H x N) – H denoting the number of patient test studies and N denoting the number of clinical features.
Medical imaging data
For each patient study use standard “four view” views (LCC, LMLO, RCC, RMLO) and discard other, nonstandard, views. If multiple images exist for the same side-view, for example, more than one LCC images in a study, use the latest image.
Step b: Feature Selection
Use the same T top selected features which were found in Training step (b), and build features matrix named “ClinicalFeaturesMat_TOP” of shape (H, T). H denoting the number of patient test studies and T denoting the number of top selected features
Step c: Infer using trained deep learning models
Infer using trained deep learning models. Use the model selected in the training, Customized IncResNetV2.
Step d: Patient Vector Representation
Build “PatientBreastSideEncoding” matrix exactly as described in training step (d).
Step e: Final Model Inference
SET final_gb_model to load trained gradient boost model from training step e. SET predictions to [] For each patient study left_side_pred <- final_gb_model(entry from “PatientBreastSideEncoding” for left breast) right_side_pred <- final_gb_model(entry from “PatientBreastSideEncoding” for right breast) final_study_pred = max(left_side_pred, right_side_pred) predictions.add(final_study_pred) #predictions now contain your final predictions over all tested patient studies.
