JB
Juan Bernabe Moreno
Director IBM Research Europa / Ireland and UK
Working across two locations (Daresbury and Hursley), our teams have been constantly contributing to creating what’s next in computing. Our research is motivated by today’s most pressing challenges both in the global context and specific to UK industries and institutions. We bring cutting-edge computational science and engineering to define the quantum computing of the future, the next generation of artificial intelligence, and the technologies that will help accelerate scientific discovery, for UK and beyond.
We are a growing team of researchers, computational scientists, engineers, and designers pioneering scientific breakthroughs. Our work advances artificial intelligence, climate and life sciences, quantum computing, and hybrid cloud.
Mathematics and algorithms for identifying configurations of complex physical systems exhibiting unique, anomalous properties.
An automated explainable bioinformatics and AI workflow for multi-omic, climate and environmental data, applied to sustainability problems e.g., nature-based carbon capture.
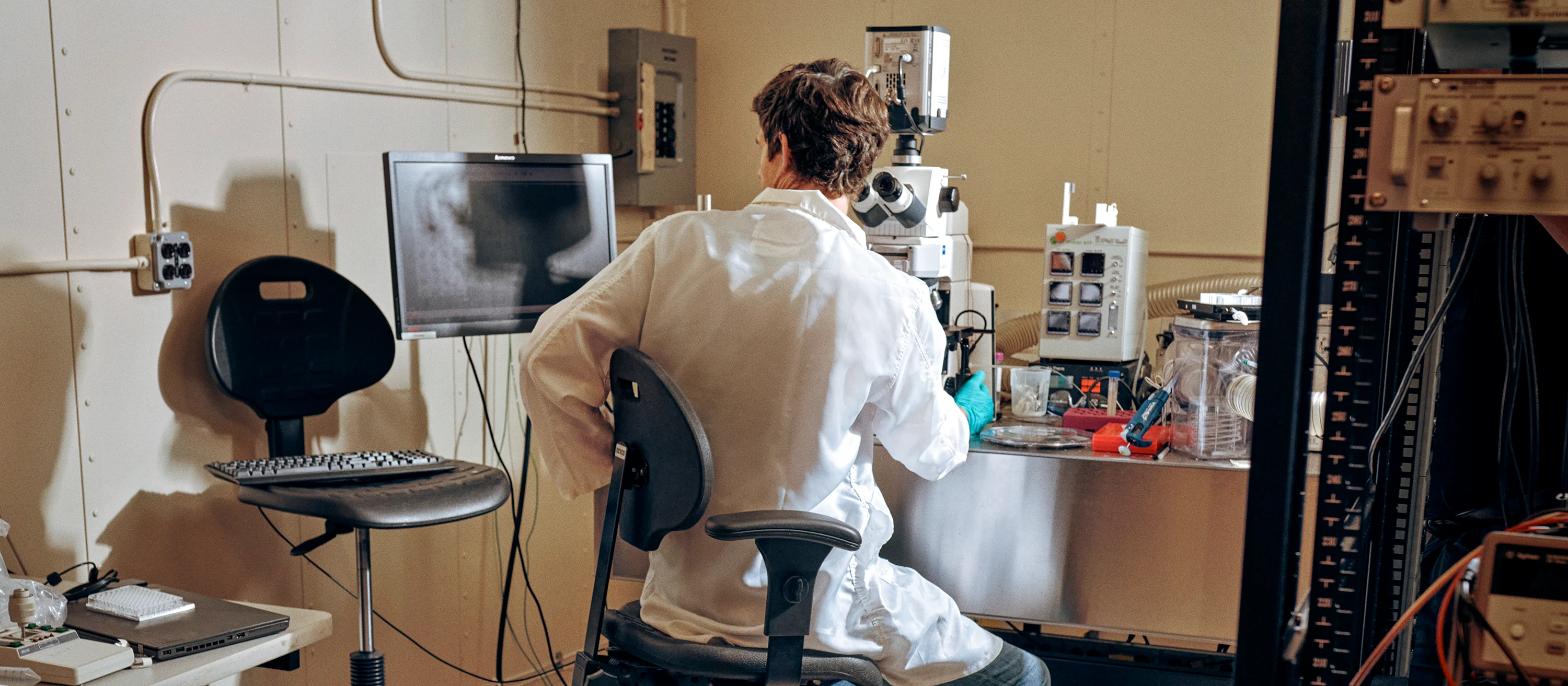
An automated explainable bioinformatics and AI workflow for multi-omic, clinical and experimental data, applied to healthcare and drug discovery problems.
The role of the Global Think Labs is to make what’s next in computing actionable for our clients. We offer a stimulating environment to explore future technologies and their transformational impact on business and society. Our Think Lab is located at the historic IBM Hursley Laboratory that brings together cutting edge IBM integration development with research into Accelerated Discovery, Quantum Computing, AI and Hybrid Cloud. Together with our colleagues from the Daresbury Lab we connect our deep technological understanding and fundamental research directly to IBM clients from around the world.
IBM United Kingdom Limited
Hursley Park
WinchesterHants SO21 2JN
UK
IBM Research
The Hartree Centre STFC Laboratory
Sci-Tech Daresbury Warrington
Warrington WA4 4AD
UK
We’re always looking for people excited to make a difference. See our open positions and help us invent what’s next.







